基于密度泛函理论(DFT)推导的能量学的微动力学模型对于解决催化的基本问题非常重要。然而,微动力学模型(MKMs)的定量保真度常常不足以最终推断出特定催化系统的机理细节。这可以归因于许多因素,如进行DFT计算的活性位点模型不正确,假设的反应机理存在缺陷,对反应条件下的表面环境考虑不足,以及DFT交换相关函数的内在错误。通过可靠的参数估计来实现实验结果与模型结果之间的一致性,并确保DFT计算与MKM预测之间的覆盖范围一致性,有可能系统地完善机理模型,从而更好地了解催化活性位。
有鉴于此,威斯康星大学麦迪逊分校的Manos Mavrikakis等人,将计算模型与反应动力学实验相结合,阐明了催化活性位点的原位性质。
本文要点
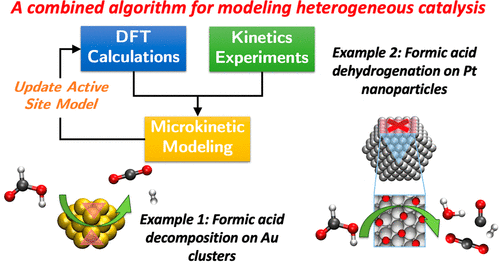
1)一般方法包括为给定的活性部位开发从头算的MKM,然后重新估计过渡态和中间态的能量,以使模型预测与反应动力学实验中测得的量匹配。如果(i)模型实验的均等性很高,则(ii)在标准DFT交换相关函数的误差范围内,合理化针对给定活性位点模型对DFT衍生的能量的调整,以及(iii)生成的MKM如果预测与用于初始化MKM的DFT计算中假定的表面覆盖率一致,可以正确识别活性位点和反应机理。如果不满足这些要求中的一项或多项,将通过更新关于活性站点结构的假设和/或通过合并覆盖效应来迭代地完善模型,直到获得高保真覆盖自洽的MKM,并最终获得动力学和热力学参数在DFT值的误差范围内。
2)以在过渡金属催化剂上甲酸(FA,HCOOH)分解的催化反应为例,介绍了如何应用该算法研究粉末金/SiC和Pt/C催化剂上的化学反应。对于仅通过脱氢反应(HCOOH→CO2+H2)发生FA分解的Au催化剂,采用了从(111)面开始迭代优化模型的方法,直到发现Au的特定集合亚纳米簇中存在的原子可以描述这种催化的活性位点。对于Pt催化剂,其中脱氢(HCOOH→CO2+H2)和脱水(HCOOH→CO+H2O)反应均具有活性,该方法确定了部分被CO*覆盖的(111)表面是活性位点,并且CO*辅助步骤大大促进了FA的总体分解活性。
参考文献:
Saurabh Bhandari et al. Combining Computational Modeling with Reaction Kinetics Experiments for Elucidating the In Situ Nature of the Active Site in Catalysis. Acc. Chem. Res., 2020.
DOI: 10.1021/acs.accounts.0c00340
https://doi.org/10.1021/acs.accounts.0c00340

















