在过去的几十年中,基于量子力学(QM)计算的原子模拟已进入化学家的工具箱,促进了对从结构表征到反应性测定的各种化学问题的理解。由于QM计算固有的低伸缩性和高计算成本,在执行大规模原子模拟时必须牺牲精度或时间。在精确度和速度之间找到一个更好的折衷点是新理论方法发展的核心。
有鉴于此,复旦大学刘智攀教授、Cheng Shang等人,综述了用于原子模拟的机器学习方法在三个关键方面的最新进展,即代表数据集的生成、ML模型的广泛性和数据表示的连续性。
本文要点
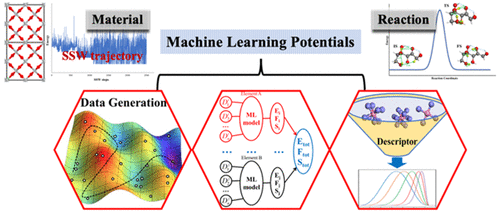
1)选取了G-NN电势在材料和反应模拟中的两个最新应用,以说明基于ML的原子模拟如何帮助发现新材料和反应。
2)基于机器学习(ML)的大规模原子模拟的最新进展显示出巨大的前景,对化学研究的多个方向均具有重要作用。基于ML的模拟不是直接求解Schrödinger方程,而是依靠大量具有准确势能面(PES)和复杂数值模型的数据集来预测总能量。这些模拟具有计算大型系统的高速和高精度的特点。由于数值模型缺乏物理基础,因此ML模型的可预测性和鲁棒性较差,而这对于应用至关重要。在建立代表性数据集时,采用全局优化方法是很自然的选择,而随机表面行走方法可以为PES上的极小区域和过渡区域提供所需的PES采样。
3)当前的ML模型通常利用局部几何描述符作为输入,并将总能量视为原子能量的总和。数据描述符和ML模型有很多形式,但材料和反应预测的应用仍然有限,尤其是因为难以训练相关的庞大全局数据集。研究表明,最近设计的幂类型结构描述符和前馈神经网络(NN)模型与高度复杂的全局PES数据兼容,这为全局神经网络(G-NN)带来了很大的潜力。
参考文献:
Pei-Lin Kang et al. Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration. Acc. Chem. Res., 2020.
DOI: 10.1021/acs.accounts.0c00472
https://doi.org/10.1021/acs.accounts.0c00472

















