吸附剂-表面相互作用的量化是一个多世纪以来非常活跃的研究领域,其重要性不可低估。例如,吸附理论有助于合理的催化剂设计,理解腐蚀,并且有助于了解表面的哪些特征会影响吸附性能。然而,发展能够将不同材料的结构和组成与其吸附性能联系起来的理论仍然是一个关键且尚未解决的挑战。建立物理上透明且定量准确的模型,以将被吸附物和固体表面之间的化学相互作用(化学吸附强度)与吸附部位的几何形状相关联,对于对催化,腐蚀和电化学的理解至关重要。
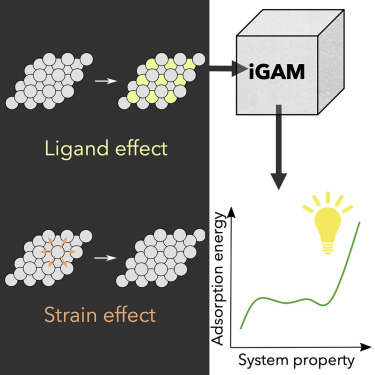
有鉴于此,密歇根大学Suljo Linic、Bryan R. Goldsmith等人,开发了一种理论指导的机器学习(ML)方法,该方法使用称为广义可加模型(iGAM模型)的一类可解释的ML模型,以发现可以量化和解释吸附位点的几何结构和化学吸附强度之间联系的可预测的结构-性质模型。
本文要点
1)采用一类可解释的机器学习模型,称为广义可加模型(iGAM),以解释和量化合金诱导的对亚表面金属合金吸附性能的变化。这些iGAM模型有助于全面了解合金局部化学环境的结构和组成变化如何影响其吸附性能。
2)通过几个案例研究来证明了这一方法,在这些案例中,分析了在不同应变和配体诱导的局部几何结构变化下,在金属合金亚表面上化学吸附的不同吸附物(O、OH、S和Cl)的化学吸附强度。
3)通过将ML衍生的化学吸附模型与先前建立的电子结构模型进行比较,阐明了控制金属表面化学吸附过程的关键物理参数。
参考文献:
Jacques A.Esterhuizen et al. Theory-Guided Machine Learning Finds Geometric Structure-Property Relationships for Chemisorption on Subsurface Alloys. Chem, 2020.
DOI: 10.1016/j.chempr.2020.09.001
https://doi.org/10.1016/j.chempr.2020.09.001

















