锂金属电池以其较高的理论容量和较低的电化学电位在电动汽车领域受到越来越多的关注。但其固有的缺点是不可控的锂枝晶生长,高化学活性以及体积变化大等严重阻碍了锂金属负极的大规模应用。近年来,研究人员已经使用各种计算研究来提高实验观察结果的合理化。
有鉴于此,苏州大学晏成林教授,钱涛综述了锂金属电池中关于分子动力学模拟的研究进展。
文章要点
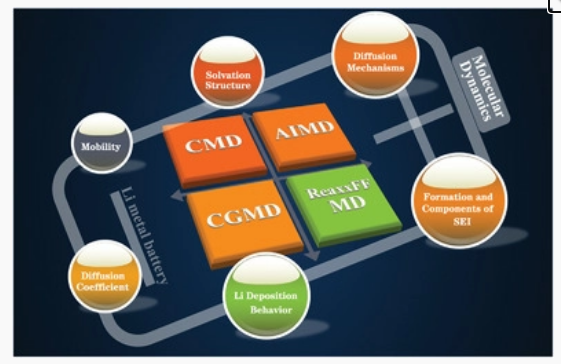
1)作者从Li+在电解液中的扩散和溶剂化结构、电极/电解液界面上Li+的形成和扩散以及不同条件下的Li沉积行为三个方面总结了不同类型的多尺度模型。
2)总结了LMB中的模拟方法,以加深对电解质配方的优化、SEI形成的机理理解和模型以及Li沉积行为的深入了解。
3)作者最后对分子动力学模拟在锂金属电池中的进一步应用提出了一些建议。
Yawen Sun, et al, Boosting the Optimization of Lithium Metal Batteries by Molecular Dynamics Simulations: A Perspective, Adv. Energy Mater. 2020
DOI: 10.1002/aenm.202002373
https://doi.org/10.1002/aenm.202002373


















