氧化铟作为一种很有前途的CO2加氢制甲醇催化剂,因其高的甲醇选择性而引起了广泛的关注。了解氧化铟催化剂的构效关系对于优化设计氧化铟催化剂具有重要的指导意义。
有鉴于此,丹麦技术大学Jens K. Nørskov等人,通过将密度泛函理论计算与微动力学建模相结合,系统地研究了In2O3(111)和In2O3(110)上的甲醇合成。
本文要点
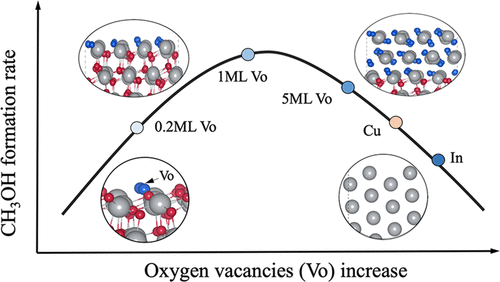
1)同时考虑了In2O3(111)和In2O3(110)表面。计算得到的表面相图表明,在实验条件下,In2O3表面最上面几层不存在晶格氧。研究了氧空位的形成能,以研究在实验条件下,随着氧空位浓度的变化,In2O3表面的稳定性。
2)结合微观动力学模型,研究了表面还原度与In2O3表面活性之间的关系,并开发了In2O3催化剂的结构-活性火山模型。还原的表面In层数与In2O3(111)的催化活性之间存在明显的关系,最佳还原层为一到两层。
3)进一步解释了为什么可以通过添加ZrO2载体来调节还原层数来优化In2O3催化剂的甲醇形成活性。
总之,该工作解释了纯In2O3催化剂的低活性,并提供了如何提高活性的理论见解。
参考文献:
Ang Cao et al. Relations between Surface Oxygen Vacancies and Activity of Methanol Formation from CO2 Hydrogenation over In2O3 Surfaces. ACS Catal., 2021.
DOI: 10.1021/acscatal.0c05046
https://doi.org/10.1021/acscatal.0c05046

















