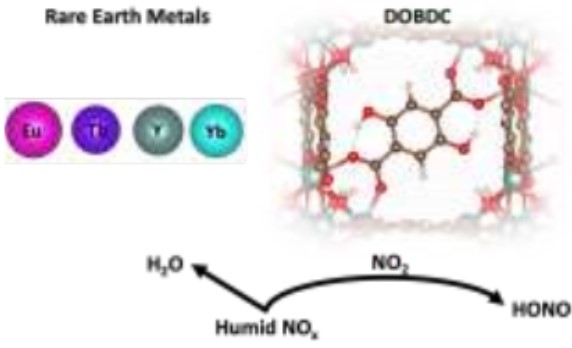
金属-有机骨架孔隙中活性气体生成是已知的骨架破坏机制;了解这些机制用作未来的耐久性设计是下一代吸附剂的关键。有鉴于此,美国桑迪亚国家实验室的Tina M. Nenoff等研究人员,开发出了用从头算分子动力学预测稀土MOFs反应性亚硝酸生成。
本文要点
1)研究人员使用了一套广泛的从头算分子动力学模拟(AIMD),首次用于预测混合酸性气体(NO2和H2O)的竞争性吸附和一系列稀土(RE)‐DOBDC MOFs的孔内反应机理。
2)研究人员确定了亚硝酸(HONO)的自发形成是由于MOF有机连接子DOBDC的去质子作用。
3)独特的DOBDC对金属杂合物的配位允许质子在没有H2O存在下,从连接物转移到NO2,可能是DOBDC MOF耐久性的一个因素。这是之前未报道过的MOFs中HONO的形成机制。
本文研究表明,利用所提出的方法可以预测未来气体在新型纳米孔材料中的相互作用。
参考文献:
Tina M. Nenoff, et al. Prediction of Reactive Nitrous Acid Formation in Rare Earth MOFs via ab initio Molecular Dynamics. Angewandte Chemie, 2021.
DOI:10.1002/anie.202102956
https://onlinelibrary.wiley.com/doi/10.1002/anie.202102956

















