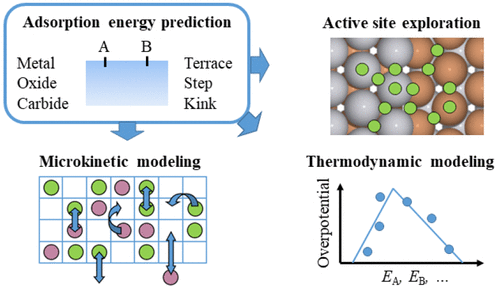
多相催化剂是一种非常复杂的材料,有许多种类(例如,金属,氧化物,碳化物)和形状。同时,催化剂表面与合成气(CO和H2)等相对简单的气相环境之间的相互作用,可能已经产生了从原子到复杂分子的多种反应中间体。创建潜在催化剂材料表面覆盖率或化学活性预测图的起点是可靠地预测所有这些中间体的吸附焓。对于简单系统,直接密度泛函理论(DFT)计算是目前的选择方法。然而,对复杂材料和反应网络的更广泛的探索通常需要以更低的计算成本度吸附焓进行预测。
有鉴于此,丹麦奥胡斯大学Mie Andersen等人,对最近的基于 ML 的方法进行分类和比较,特别关注模型稀疏性、数据效率以及人们可以从模型中获得的物理信息。
本文要点
1)利用机器学习(ML)和相关技术对量子力学计算进行准确和低成本的预测,近来得到了越来越多的关注。所采用的方法从物理动机模型到混合物理- ΔML方法再到完整的黑盒方法,如深度神经网络。在最近的工作中,他们探索了使用压缩感知方法(Sure independent Screening and Sparsifying Operator, SISSO)来识别稀疏(low-dimensional)描述符的可能性,用于预测金属和氧化物的各种活性位点上的吸附焓。从一组物理上的主要特征开始,比如原子的酸/碱性质、配位数或带矩,然后让数据和压缩感知方法找到这些特征的最佳代数组合。
2)虽然迄今为止的许多工作仅专注于对吸附焓等数据库的预测,但也出现了一个新的兴趣,即开始使用ML预测来回答有关多相催化剂或非均相催化剂功能的基础科学问题,甚至可能设计出比我们今天所知的更好的催化剂。在使用基于比例关系的模型(火山曲线)的工作中,这项任务得到了显着简化,其中模型结果仅由一个或两个吸附焓决定,因此成为基于 ML 的高通量筛选或设计的唯一目标。然而,廉价的ML能量学的可用性也允许超越比例关系。
参考文献:
Mie Andersen et al. Adsorption Enthalpies for Catalysis Modeling through Machine-Learned Descriptors. Acc. Chem. Res., 2021.
DOI: 10.1021/acs.accounts.1c00153
https://doi.org/10.1021/acs.accounts.1c00153

















