scHi-C可以识别细胞间三维(3D)染色质组织的变异性,但相互作用的稀疏性构成了分析的挑战。有鉴于此,美国卡内基梅隆大学的Jian Ma等研究人员,实现多尺度和综合单细胞Hi-C分析。
本文要点
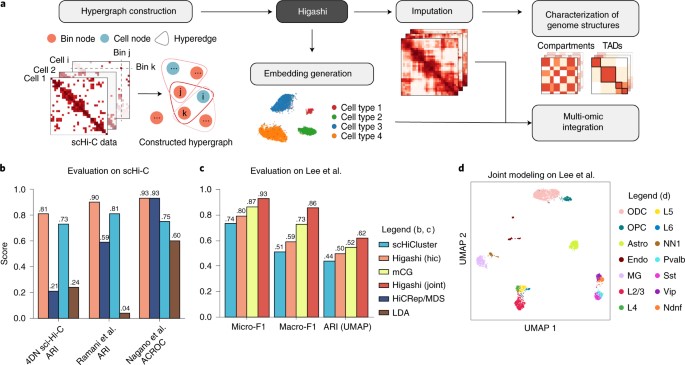
1)研究人员报告了Higashi,一种基于超图表示学习的算法,可以将单细胞之间的潜在相关性纳入其中,从而加强接触图的整体归纳。
2)Higashi优于现有的单细胞Hi-C(scHi-C)数据的嵌入和估算方法,能够识别单细胞中的多尺度三维基因组特征,如区室化和TAD样域边界,能够细化划定其细胞间的变化。
3)此外,与单独分析两种模式相比,Higashi可以将同一细胞中联合剖析的表观基因组信号纳入超图表示学习框架,从而改善单核甲基3C数据的嵌入。
4)在人类前额叶皮层的scHi-C数据集中,Higashi确定了三维基因组特征和细胞类型特定基因调控之间的联系。
本文研究表明,Higashi也有可能被扩展到分析单细胞多向染色质相互作用和其他多模态单细胞全向数据上。
参考文献:
Ruochi Zhang, et al. Multiscale and integrative single-cell Hi-C analysis with Higashi. Nature Biotechnology, 2021.
DOI:10.1038/s41587-021-01034-y
https://www.nature.com/articles/s41587-021-01034-y

















