电化学活化势垒的准确理论模拟是理解电催化和指导更有效催化剂设计的关键。提供质子转移过程的详细过程会遇到几个挑战:电荷转移过程中的恒定电位要求、过程中涉及的不同时间尺度以及溶剂的热波动。因此,应用密度泛函理论 (DFT) 计算来模拟电极-溶剂界面处的电位相关活化势垒,其计算成本非常昂贵,而且结果令人怀疑。
有鉴于此,美国SLAC国家加速器实验室Frank Abild-Pedersen等人,开发了一种基于电荷守恒和解耦势能表面的分析方法来计算电荷转移势垒。
本文要点
1)该方法是基于质子给体和质子受体的解耦势能面(PESs)的PET步骤。通过定义两个 PESs的耦合强度,从反应的组合绝热 PESs中获得了PET 步骤的势垒。同时,这严格遵守电荷守恒。
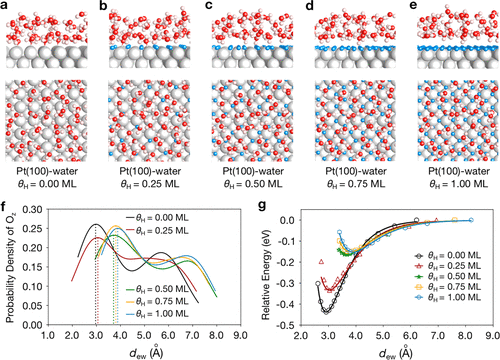
2)该方法可以模拟不同电位下的电化学过程,并明确包括电极-溶剂界面处溶剂的热波动。使用 Pt 催化的碱性析氢反应 (HER) 作为基准反应,并在考虑室温下金属表面和第一溶剂层之间的空间波动的情况下对 HER 的微动力学进行建模。
3)水-金属距离的分布对电荷转移过程的势垒有很大影响,与从静态溶剂转移相比,反应网络的统计波动导致HER电流增加了几个数量级。
总之,该工作中提出的模型成功地模拟了 HER 中不同反应机制的趋势,获得的理论 I-V 曲线与实验结果具有良好的定性一致性。
参考文献:
Jiang Li, et al. Modeling Potential-Dependent Electrochemical Activation Barriers: Revisiting the Alkaline Hydrogen Evolution Reaction. J. Am. Chem. Soc., 2021.
DOI: 10.1021/jacs.1c07276
https://doi.org/10.1021/jacs.1c07276

















