在设计新型多相催化剂时,阐明反应机理以及活性位点本身的几何与电子结构是一项极具挑战性的重要任务。这种深度研究最好通过一种包括环境压力催化、表面科学和理论预测的多管齐下的方式来进行。
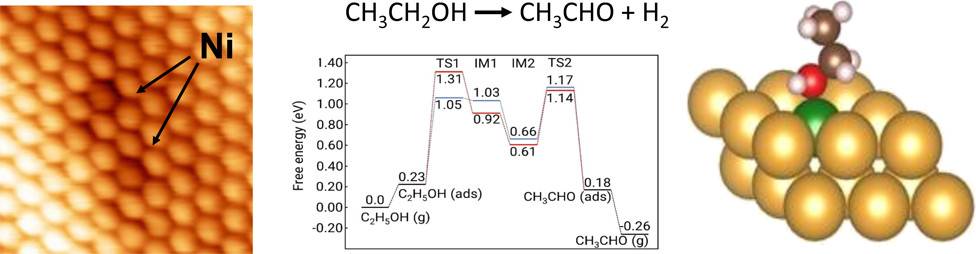
近日,SLAC国家加速器实验室Simon R. Bare,加州大学洛杉矶分校Philippe Sautet,塔夫茨大学E.Charles H.Sykes报道了作者利用上述策略深入研究了NiAu单原子合金(SAA)催化剂在乙醇选择性非氧化脱氢(EDH)制乙醛和氢气反应中的机理。
文章要点
1)Ni的原子级分散对于乙醇至乙醛的选择性转化至关重要,即便Au表面存在很小的Ni簇,也会通过C–C断裂而形成不良副产物。对反应机理的光谱、动力学和理论研究表明,C–H和O–H键断裂步骤在动力学上是相关的,并且单原子Ni被确认为活性中心。
2)X射线吸收光谱研究可以追踪Au主体中Ni原子在反应前、反应中和反应后的电荷。具体而言,在初始状态下,Ni原子携带部分正电荷,其在乙醇中与电负性氧配位时增加,在解吸时减少。这种氧化态循环类似于单位点均相催化剂的行为。
3)考虑到许多单位点催化剂都具有独特的电子结构,这种组合型方法可以根据其反应环境监测单原子位点的原子级催化剂结构和电荷状态,是研究结构-功能关系的关键步骤,有望为新催化剂的设计提供指导。
参考文献
Georgios Giannakakis, et al, Mechanistic and Electronic Insights into a Working NiAu Single-Atom Alloy Ethanol Dehydrogenation Catalyst, J. Am. Chem. Soc., 2021
DOI: 10.1021/jacs.1c09274
https://doi.org/10.1021/jacs.1c09274


















