锂-硫(Li-S)电池(LSBs)因其低成本和高硫含量而成为最有前途的储能技术之一。然而,多硫化物穿梭及其相应的容量衰减问题是其商业化的主要障碍。利用固态电解质(SEs)代替传统的液态电解质是一种实现LSBs商业化潜在的解决方案。
近日,印度理工学院Swastika Banerjee,加州大学圣地亚哥分校Shyue Ping Ong使用密度泛函理论(DFT)计算和机器学习原子间势,对全固态LSB中正极-电解质界面的热力学和动力学进行了全面的研究。
文章要点
1)发现,在主要的固体电解质化学物质(氧化物、硫化物、氮化物和卤化物)中,硫化物SE通常对S8正极最稳定,而其他SE化学物质则具有高电化学不稳定性。如果出于其他原因需要使用其他SEs化学物质,则几种二元和三元硫化物(例如,LiAlS2、Sc2S3、Y2S3)是优异的缓冲层。
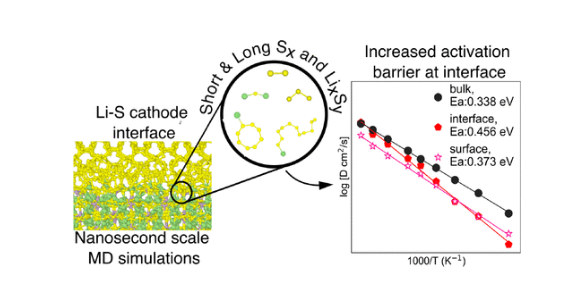
2)研究人员使用主动学习方法发展了一个精确的矩张量势来研究S8|β-Li3PS4界面。大界面模型(> 1000s原子)的分子动力学(MD)模拟显示,最稳定的Li3PS4(100)表面倾向于与S8形成界面,具有2D通道和较低的Li扩散激活势垒。
这些结果为下一代全固态LSB的正极-电解质界面设计提供了重要的新见解。
参考文献
Thermodynamics and Kinetics of the Cathode−Electrolyte Interface in All-Solid-State Li−S Batteries Manas Likhit Holekevi Chandrappa, J. Am. Chem. Soc., 2022
DOI: 10.1021/jacs.2c07482
https://doi.org/10.1021/jacs.2c07482


















