金属卤化物钙钛矿是用于未来光电子应用的有前景的材料。对于许多应用来说,一个有趣的特性是通过成分工程实现带隙的可调性。虽然关于吸收或光致发光变化的实验报告显示出对不同化合物的具有高度的一致性,但这些变化的物理起源,即化合价和导带位置的变化,尚未得到很好的表征。近日,埃因霍温理工大学Shuxia Tao联合科隆大学
Selina Olthof使用光电子能谱数据确定所有初级锡和铅基钙钛矿的电离能和电子亲和力值,由第一性原理计算和紧束缚分析支持。研究人员证明能级变化主要取决于金属阳离子和卤素阴离子的原子能级的相对位置,其次受阳离子 - 阴离子相互作用强度的影响。这些结果朝着理解该类材料的电子结构迈出了重要的一步,并为钙钛矿光电子学中关于能量学的合理设计规则提供了基础。
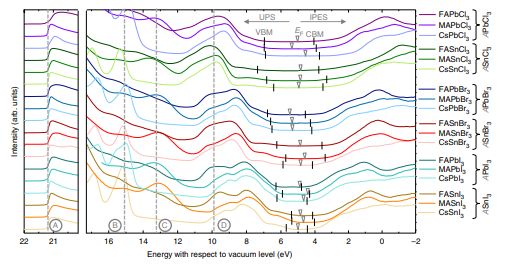
Tao, S. Olthof, S. et al. Absolute energy level positions in tin- and lead-based halide perovskites. Nat. Commun. 2019.
DOI:10.1038/s41467-019-10468-7
https://www.nature.com/articles/s41467-019-10468-7


















